
Summer 2014 - Vol. 9, No. 2
Familial Hypercholesterolemia
Joseluis Ibarra, MD, FACC
The Preventive Cardiology and Apheresis Clinic,
Lancaster Vascular and Heart Institute
INTRODUCTION
This year the Preventive Cardiology and Apheresis Clinic of The Lancaster Vascular and Heart Institute will begin the Familial Hypercholesterolemia Initiative in this community. With more comprehensive screening for hypercholesterolemia, we hope to achieve earlier identification and treatment of this disease and prevent its devastating sequelae.
Familial Hypercholesterolemia (FH) is a common genetic disease that causes premature and aggressive coronary artery disease (CHD) and death. Presence of the causative genetic defect increases the risk of CHD 20-fold and is the cause of up to 5% of heart attacks before the age of 60 and 20% before the age of 45.1-3 Unfortunately, most diagnoses are made only after a cardiac event; at best, efforts to screen and detect for primary prevention have been weak. In 2011 the National Lipid Association began a more proactive population screening effort. It is the goal of the Vascular and Heart Institute to provide the same benefit to Lancaster County families.
DEFINITION AND PREVALENCE
The Familial Hypercholesterolemias are a group of inherited autosomal dominant genetic defects with more than 90% gene penetrance. These result in very high elevations of serum cholesterol and the subsequent consequences of severe and premature coronary artery disease (CHD).4 Once thought to be a single genetic abnormality affecting only the low density lipoprotein receptor(LDLR), there are new and ongoing discoveries of defects in other key genes involving apolipoprotein B (ApoB), protein convertase subtilsin/kexin type 9 (PCSK9), low-density lipoprotein receptor adapter protein 1 (LDLRAP1), and others yet to be defined.5 Each of these entities alone can lead to severe hypercholesterolemia. More concerning is that the combination of several defects in the same individual can certainly magnify the clinical expression, worsen outcomes, and make management more difficult.
The homozygous form (HoFH) is fortunately quite rare, occurring in approximately 1 out of every 1,000,000 individuals who inherit the genetic defect from both parents. The defect or defects leads to hypercholesterolemia in childhood with LDLs that range from 600 to >1000 mg/dL, with early development of CHD. These children can develop eruptive xanthomas in their Achilles tendons, wrists, knees and gluteal areas as early as 8 months of age. They subsequently develop coronary and peripheral vascular disease including myocardial infarction and strokes; they undergo coronary and peripheral bypass procedures (stents, bypass grafts) in their late childhood and early teens; and they succumb in their second or third decade of life.6,7 Heterozygous FH (HeFH) occurs in 1 in 300-500 individuals in the general population, though at higher rates (1 in 100) in certain “Founder Groups.” These include French Canadians, Christian Lebanese, South African Afrikaaners, Asian Indians, and Ashkenazi Jews. Although these individuals inherit the genetic defect from only one parent, the gene penetrance is as profound as that seen with the homozygotes. Their LDL cholesterol typically ranges from 350 to 550 mg/dL but, unlike the homozygotes, their onset of clinical presentation can be delayed for 10-20 years. Approximately 50% of men and 30% of women with this disorder will have fatal or non-fatal coronary events before ages of 50 and 60 respectively. The mean onset of events is usually in the early 40s for men and early 50s for women.8,9
There are approximately 10 million FH patients worldwide, 625,000 patients in the United States, and >2800 in Lancaster County alone.
GENETICS
FH is due to mutations in the LDLR gene including deletions, missense, nonsense, and insertion types. There are over 1600 mutations of the LDLR gene which account for 85-90% of cases of FH. Patients with the defect have either no LDL receptors or ineffective ones that have reduced affinity for LDL. The resulting abnormality involves an inability of the receptor to clear LDL-cholesterol from the circulation.10
LDLR defects are classified into 5 types based upon the physiologic consequences of the defect:11
- Class I: LDL receptor is not synthesized at all.
- Class II: LDL receptor is not properly transported from the endoplasmic reticulum to the Golgi apparatus for expression on the cell surface.
- Class III: LDL receptor does not properly bind LDL on the cell surface because of a defect in either apolipoprotein (Apo) B-100 (R3500Q) or in the LDL receptor.
- Class IV: LDL receptor bound to LDL does not properly cluster in clathrin-coated pits for receptor-mediated endocytosis.
- Class V: LDL receptor is not recycled back to the cell surface.
ApoB mutations are termed Familial Defective ApoB (FDB) and are less severe than the LDLR mutations. The most common is the Arg35000Gln and can account for up to 5-10% of FH.12
Protein convertase subtilisin/kexin type 9 (PCSK9) defects tend to increase LDL receptor degradation which leads to less “active” LDL receptors, thus increasing the amount of circulating LDLs available for oxidation and subsequent plaque formation.13
Other non-dominant mutations include LDLRAP1, sitolsterolemia, and deficiency of cholesterol 7-alpha hydroxylase (CYP7A1).14
DIAGNOSIS
The diagnosis of Familial Hypercholesterolemia is determined by a combination of factors in the evaluation of a patient, including medical history, family history, physical examination, and laboratory findings. Criteria that have been established and validated include the Simon Broome Register Diagnostic Criteria (App available),15 the Dutch Lipid Clinic Network Diagnostic Criteria,16 and the U.S. Make Early Diagnosis Prevent Early Death (MEDPED) Program Diagnostic Criteria for FH.17
CONSIDERATIONS FOR DIAGNOSIS.18
- Patient's prior history of CHD, age of onset, current cardiovascular symptoms (if any), use of lipid-lowering agents, and history of the dyslipidemia’s response to medication.
- A family history of premature CHD in first degree relatives (males < 55 and or females < 65).
- A History of severe hyperlipidemia in the family; particularly if there is a bimodal distribution of LDL in siblings with unaffected members having LDLs of 130 mg/dL or less and affected members having at least two-fold higher values of >190-220 mg/dL.
- Physical findings, though insensitive, are quite specific; tendon xanthomas are pathognomonic for FH but unfortunately occur in less than half of patients. These should be sought by both inspection and palpation to ascertain their presence and particularly in the Achilles tendon (most common), finger extensors, and - least often - patellar tendons. Tuberous xanthomas and xanthelasma in younger patients are non-specific but suspicious findings that warrant further evaluation. It is important to note is that the absence of any of these physical findings does NOT exclude the diagnosis of FH.
- The diagnosis is confirmed if a family member is found to have a tendon xanthoma and concomitant high cholesterol values.
- Secondary causes of the dyslipidemia such as undiagnosed diabetes mellitus, primary biliary cirrhosis, nephrotic syndrome and hypothyroidism should be excluded.
Nonetheless, genetic testing is considered the "gold standard." If a mutation has already been identified in a family group, it can give definite confirmation of the presence of a causal mutation in the LDLR, APOB, or PCSK9 genes. However, the testing is cost prohibitive, and in patients with possible FH, the identification rate is only about 50%. Identification is even more limited by the ongoing discovery of new gene defects that remain incompletely studied and classified. It is important to remember that the lack of identification by genetic testing does not exclude FH.
ADULT TREATMENT OF FAMILIAL HYPERCHOLESTEROLEMIA
Adult patients (age> 20) with LDL cholesterol ≥ 190 (non-HDL ≥ 220) require early and aggressive management with a goal of reducing LDL 50% or more, to decrease the high lifetime risk of CHD.
Since there are differences in the traditional risk factors for patients with FH, the traditional risk factor algorithms do not apply and should not be used in this patient population.19
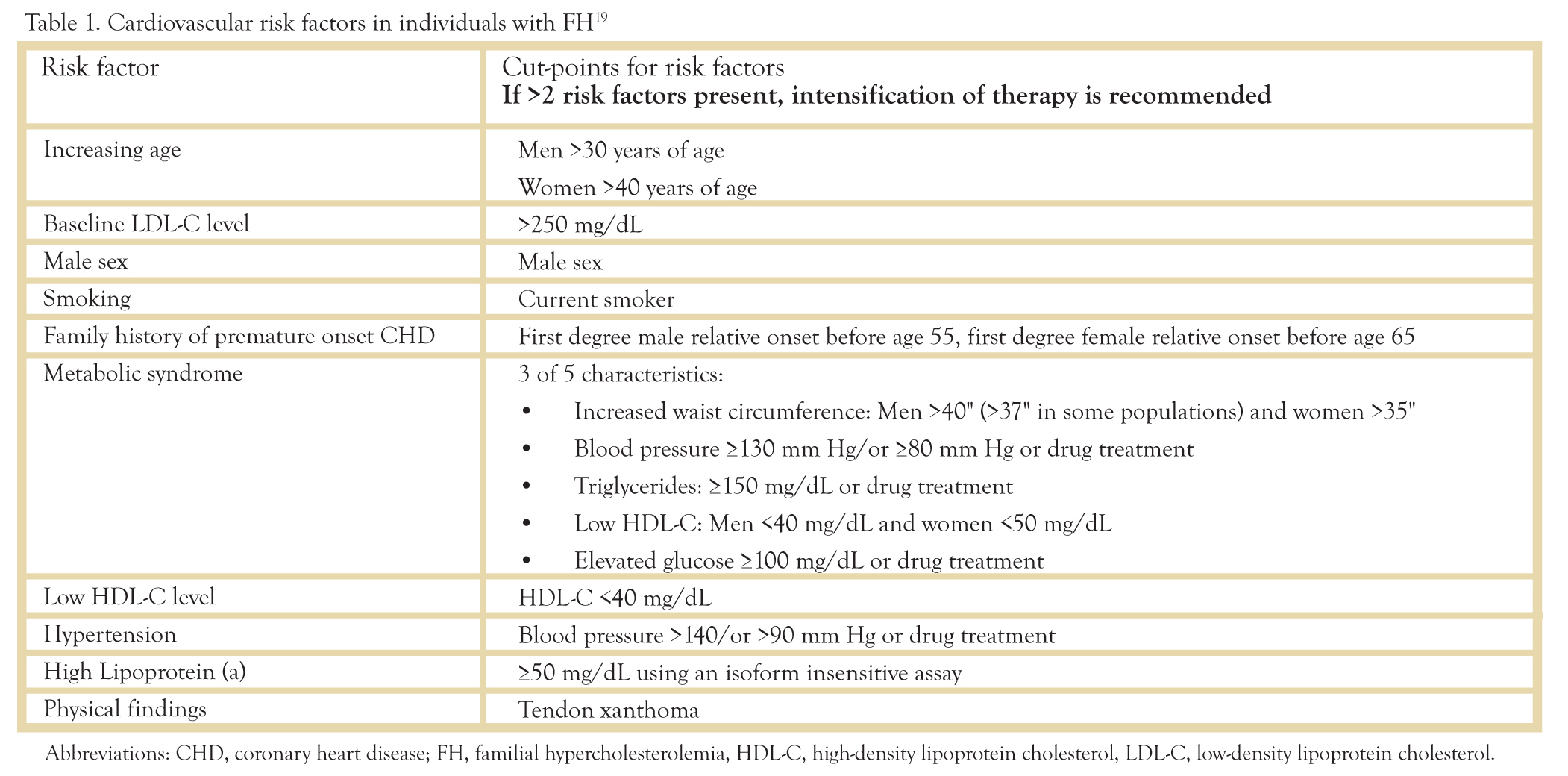
Table 1: Cardiovascular risk factors in individuals with FH.19
In higher risk patients, it is necessary to intensify treatment to reach goals of an LDL < 100 and non-HDL < 130. This includes individuals with already established CHD/CVD (MI, TIA/CVA, PAD, resuscitated cardiac arrest, PCI, CABG, angina pectoris, carotid artery stenosis >50%, abdominal aortic aneurysm, diabetes mellitus, family history of premature coronary artery disease, ongoing smoking, elevated Lp(a) > 50 mg/dL, and two or more FH risk factors.
Non-pharmacological management such as diet and weight control, exercise, and particularly smoking cessation should be the cornerstone of management but should not be a substitute for, nor delay the early onset of drug therapy. Smoking is very concerning as it tends to accelerate the atherosclerotic disease process in patients with FH, particularly in men. Thus a more aggressive smoking cessation program and/or pharmacological agents may be required. Attempts should be made to maintain optimum blood pressure control < 140/90 (<130/80 in diabetic patients) and aspirin (75-81 mg daily) should be considered for those at high risk of CHD or CVA.
All contemporary guidelines recommend statins as the first line agents for management of FH. Though there are no large randomized control studies assessing outcomes in management of FH, Dutch studies and analyses of the UK Simon Broome database have shown marked improvement in lipid levels and a significant reduction of risk when compared to levels of non-FH patients. The risk improvement was most notable with early institution of statins and in patients with the highest lipid values at baseline.
In patients who do not achieve the desired goal of therapy, the addition of ezetimibe, niacin, or bile acid sequestrants is reasonable if they are well tolerated and the cost does not lead to decreased adherence.19
Overall, statins alone or in combination with other lipid-lowering agents can reduce LDL by an average of 25% in homozygous and 45-60% in heterozygous FH patients. If these additional agents do not achieve goal, or management becomes difficult, referral to a Lipid specialist is recommended.
The Preventive Cardiology and Apheresis Clinic of the Lancaster Vascular and Heart Institute has the ability to provide recently approved registry agents to assist in the difficult management of FH patients. Among these are:
- Lomitapide, which inhibits microsomal transfer protein that is essential for very low LDL (VLDL) assembly and metabolism.20
- Mipomersen, which is an antisense oligonucleotide that results in decreased production of Apo B.21
- PCSK9 inhibitors, which prevent LDL receptor degradation and extend LDL receptor functionality.22
Finally, patients who are intolerant of any of these agents or do not achieve the necessary goals to prevent worsening cardiovascular disease, might be candidates for Lipid Apheresis. This treatment, akin to dialysis, involves plasma separation and filtering through several mechanisms with an end result of lowering LDL up to 83%, VLDL up to 70%, and Lp(a) up to 76%.
In a 2-4 hour session performed every 2-3 weeks, LDL, VLDL, and Lp(a) are removed from the plasma and the patient’s baseline elevated values are lowered to non-FH levels. Because of the ineffective LDL-R mutations that exist in these individuals, the levels gradually rise back to their pre-apheresis levels. An additional benefit with this treatment is that with time, the actual baseline levels can re-set to lower values. There are data which indicate that reductions in CV events are proportional to the degree of LDL lowering.
CASCADE SCREENING
Cascade screening is the process of identifying family members at risk for FH, and is an integral part of adult management. This involves screening all first-degree relatives of the affected patient, particularly children and young adults, and – if possible – can include 2nd and 3rd degree relatives.
In any given population it is estimated that only approximately 20% of FH patients are diagnosed and less than 10% receive appropriate treatment. As a result, cascade screening is the most efficient and cost-effective mechanism to identify those at risk. There is a 50% probability of detection in first-degree relatives, 25% in second-degree relatives, and 12.5% in third degree relatives.23
CONCLUSION
The hope of the Preventive Cardiology & Apheresis Clinic and The Lancaster Heart and Vascular Institute is to improve the cardiovascular health of this region with the aid of our colleagues, by leading this effort to identify and aggressively treat this genetic disorder.
REFERENCES
1. Goldstein, JL, Hazzard WR, Schrott HG, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. I. Lipid levels in 500 survivors of myocardial infarction. J Clin Invest. 1973;52:1533-1543.
2. Genest JJ Jr., Martin-Munley SS, McNamara JR, et al. Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation. 1992;85:2025–2033.
3. Dorsch MF, Lawrance RA, Durham NP, Hall AS. Familial hypercholesterolaemia is underdiagnosed after AMI. BMJ. 001;322:111.
4. Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259–289.
5. Hopkins PN, Toth PP, Ballantye CM, Rader DJ. Familial Hypercholesterolemia: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3suppl):S9-17.
6. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001. p. 2863–2913.
7. Kwiterovich POJr. Recognition and management of dyslipidemia in children and adolescents. J Clin Endocrinol Metab. 2008;93:4200–4209.
8. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Familial hypercholesterolemia and coronary heart disease: a HuGE association review. Am J Epidemiol. 2004;160:421–429.
9. Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatement of familial hypercholesteroleamia. Atherosclerosis. 2003; 168L1-14.
10. Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Anu Rev Genet. 1990; 24:133-170.
11. Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 992;1:445–466.
12. Varret M, Abifadel M, Rabes JP, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet. 2008;73:1–13.
13. Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–77.
14. Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4:214–225.
15. Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991;303:893–896.
16. Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55–68.
17. WHO Famlial Hypercholesterolemia Consultation Group. Familial Hypercholesterolemia. Report of a WHO Consultation. Paris: World Health Organization; 1998 October 3, 1997.
18. Hopkins PN, Toth PP, Ballantye CM, Rader DJ. Familial Hypercholesterolemia: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011; 5 (3suppl): S9-17.
19. Robinson JG, Goldberg AC. Treatment of Adults with Familial Hypercholesterolemia and evidence for Treatment: Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011; 5 (3 suppl): S18-29.
20. Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE, Rader DJ. 2007. "Inhibition of Microsomal Triglyceride Transfer Protein in Familial Hypercholesterolemia". New England Journal of Medicine 356 (2): 148–156.
21. El Harchaoui K, Akdim F, Stroes ES, Trip MD, Kastelein JJ. 2008. "Current and future pharmacologic options for the management of patients unable to achieve low-density lipoprotein-cholesterol goals with statins". Am J Cardiovasc Drugs 8 (4): 233–42.
22. Sun XM, Eden ER, Tosi I, Neuwirth CK, Wile D, Naoumova RP, Soutar AK. 2005. "Evidence for effect of mutant PCSK9 on apolipoprotein B secretion as the cause of unusually severe dominant hypercholesterolaemia". Hum. Mol. Genet. 14 (9): 1161–9.
23. Herman K, Van Heyningen C, Wile D. Cascade screening for familial hypercholesterolaemia and its effectiveness in the prevention of vascular disease. Br J Diabet Vasc Dis. 2009;9:171–174.