
Summer 2011 - Vol.6, No.2
Of Old Dogs and New Tricks:
The Epidemic of Hyper-virulent Clostridium difficile Infection
Joseph M. Kontra, M.D.
Abstract
Clostridium difficile (CD) has been a well-recognized cause of antibiotic-associated diarrheal disease since the late 1970’s. A mutation in toxin gene regulation, however, has unleashed a new epidemic of hyper-virulent infection. This new strain of CD, designated B1/NAP1/027, has spread rapidly across North America, Europe, and Asia, and has now supplanted Methicillin-Resistant Staphylococcus Aureus (MRSA) as the leading cause of nosocomial infection in some U.S. hospitals.
This paper highlights the changing epidemiologic and clinical features of this born-again pathogen, and discusses new approaches to the diagnosis, treatment, and prevention of modern era Clostridium difficile infection (CDI).
Scope and Impact of CDI
Toxin-producing strains of CD were first linked to cases of antibiotic-associated diarrhea in 1978. By the year 2000, CDI was known to cause 15-25% of cases of diarrhea associated with the use of antibiotics, particularly in hospital settings. In Quebec in 2003, an outbreak of severe diarrheal disease with high morbidity and mortality due to CD was described.1 The incidence of CDI there quadrupled, and was associated with an unusually high attributable mortality of 6.9%, compared with a baseline of 1.5%. Outbreaks in the U.S. quickly followed. Subsequent analysis of microbial samples from Quebec as well as from six U.S. east coast states demonstrated the clonal nature of the outbreak strain, subsequently dubbed the North American Pulsed Field type 1 (NAP1) strain of CD.2 Other designations for this same strain include restriction endonuclease strain B1, and PCR ribotype 027 (or simply B1/NAP1/027).
Since 1996, the incidence of CDI in the U.S. has more than doubled, with an estimated 3 million cases each year. Thus CDI in now possibly the most common identified bacterial cause of acute diarrhea in the U.S.3 The total estimated cost (in 2005 dollars) of care for patients with CDI exceeds $3.2 billion.4 In a 2010 prospective cohort study of 28 hospitals in the Duke Infection Control Outreach Network, the rate of nosocomial CDI exceeded the rate of nosocomial MRSA infection for the first time.5
Microbiology and Genesis of the Epidemic Strain
CD is a very difficult microbe to cultivate (hence the name), and requires specialized media, prolonged incubation time, and considerable expertise. It also requires a high olfactory tolerance, as this organism produces the pungent aromatic amines putrescine and cadaverine, which account for the characteristic odor associated with this organism and this disease. Cultivation of CD is typically carried out only in research facilities. Since the disease is defined by toxin production, cultivation of the organism alone is not sufficient to establish the diagnosis of CDI. Toxins A (enterotoxin) and B (cytotoxin) are the major virulence factors for CDI. Genetic regulation of toxin production resides in the pathogenicity locus, which consists of one gene for each toxin, plus three regulatory genes.
The B1/NAP1/027 strain of CD is characterized by a deletion in the tcdC repressor gene responsible for down-regulation of toxin production. This results in a greater than 10-fold increase in production of both toxins A and B. In addition, this strain also produces a unique binary toxin identical to the iota toxin of Clostridium perfringes. While the exact contribution to pathogenesis of the binary toxin remains to be elucidated, some research suggests that it may act synergistically with toxins A and B to produce the severe colitis characteristic of this endemic strain.6 The organism is also a hyper-producer of spores, which are resistant to most environmental disinfectants, and for which there is no known effective antimicrobial treatment. Moreover, B1/NAP1/027 CD is characteristically resistant to fluoroquinolones, which may provide an additional survival advantage.
Changing Epidemiology
The traditional risk factors associated with CDI infection even prior to the advent of NAP1 are well known, and include prior antibiotic exposure, hospitalization or residence in an extended care facility, advanced age, and immune suppression. The ascendency of the NAP1 strain of CD, however, has changed the epidemiologic landscape of this disease, exposing new groups of patients to potential disease.
Neonates become colonized with CD shortly after birth, and high levels of detectable toxin are characteristically present in neonates without any associated clinical disease. Delayed expression of CD toxin receptors by neonatal colonic mucosa until one year of age has been postulated as the explanation, although this remains a topic of investigation. Nonetheless, prior to the epidemic of hyper-virulent CDI, disease in children was uncommon. With the advent of NAP1, however, from 2000 to 2006 the incidence of CDI in children aged 1-17 years more than doubled.7
While the elderly have always been at risk for CDI, in the NAP1 era both incidence and mortality rates in the elderly have escalated. In those over 85 years, mortality more than doubled from 2000 to 2005.8
A unique feature of the NAP1 epidemic is the occurrence of CDI in normal, previously healthy children and adults without exposure to antibiotics or to health care facilities. Acute community-acquired CDI in peripartum women is also now well-described, including cases requiring emergency colectomy.9 An association of CDI with the prior use of gastric acid suppressing agents has been postulated.10 Thus, NAP1 CDI has begun to resemble diarrheal disease caused by traditional food-borne pathogens.
Pathophysiology
While intestinal colonization with CD is common in neonates, healthy older children and adults tend to have very low colonization rates. In contrast, patients who have been recently hospitalized have markedly elevated rates of colonization, on the order of 20-30% in hospitals and 50% in extended care facilities. Acquisition of spores from the inanimate environment in health care facilities and from the hands of health care workers is the likely explanation. Thus, a carrier state is established, contributing to ongoing environmental contamination.
Though antibiotic exposure can light the fuse for the subsequent development of CDI, host factors are now recognized to also play a major role in pathogenesis. Patients with chronic colonization with CD and high innate anti-Toxin-A antibodies tend to have a low likelihood of development of CDI after antibiotic exposure, as well as a lower probability of relapse. Conversely, patients exposed to antibiotics with low baseline anti-toxin antibody levels, and who experience a new acquisition of CD are more likely to have acute and more severe disease with a higher rate of relapse.11
Clinical Features
Diarrhea with colitis is the most common manifestation of CDI, typically in association with fever and leukocytosis. Endoscopic evaluation typically reveals pseudomembranous colitis, which is associated histopathologically with involvement of the epithelium and lamina propria. Computerized tomography (CT) characteristically reveals segmental colonic wall thickening. In more severe cases, and in particular with NAP1 infection, involvement may become pan-colonic with transmural inflammatory changes on histopathology. This fulminant colitis may lead to toxic megacolon, septic shock, and perforation, with a mortality of 45%. Total colectomy may be required as a life-saving intervention, but even this may not be curative in hyper-virulent infections. A unique feature of NAP1 CDI post-colectomy is the development of CDI of the ileum. Patients can present with sepsis, profound diarrhea, and ileitis due to a relapse of CDI.
Relapsing CDI remains a challenging problem for clinicians, as the relapse rate after the first episode of CDI is about 20%, regardless of the antimicrobial utilized. This rises to 40% after the first relapse, and 60% after two relapses.12 Randomized controlled trials are needed to establish the best course of management.
New Trends in Laboratory Diagnosis
As discussed, cultivation of CD in vitro is problematic, and not within the reach of most hospital laboratories. In addition, a positive culture may represent colonization with a strain that may or may not produce toxin.
One approach to screening stool samples is to test for glutamate dehydrogenase (GDH, or common antigen) by EIA (enzyme immunoassay). CD produces large amounts of GDH, and a stool specimen negative for GDH can be reported as negative for CD. The assay, however, detects both toxin-positive and toxin–negative organisms, and thus a stool positive for GDH must be accompanied by a separate assay for CD toxin production. This two-step approach has limited the test’s utility.
Direct toxin detection has become the most often utilized approach to the diagnosis of CDI. The gold standard traditionally has been the cell culture cytotoxicity assay, wherein a monolayer of susceptible cells is exposed to a stool filtrate, with or without the addition of a monoclonal antibody. Inhibition of cytopathic effects by specific anti-Toxin A and B antibodies establishes the presence of CD toxins. The assay is capable of detecting as little as 10 picograms of toxin, but it is time-consuming, expensive, and requires the materials and expertise of reference labs or research facilities. Furthermore, results take several days, and are thus not available in the real-time required for patient management decisions.
Toxin detection by EIA was the next step in the evolution of CDI diagnostics. Commercial EIA kits capable of detection of Toxin A and or Toxin B with a rapid turn-around time are available and cost effective, but the limit of detection of EIAs is 100-1000 picograms of toxin, which is much higher than the cytotoxicity assay. Thus, the specificity of EIA assays is high (>95%), but the sensitivity is lacking (65-90%). Unfortunately, despite what is best described as medical folklore, repeated testing of stool samples by EIA does not increase the yield significantly, as demonstrated by multiple studies.
PCR (Polymerase Chain Reaction) detection of the gene responsible for CD Toxin B production has recently become commercially available. The assay can be performed directly on a liquid stool sample and has a rapid turn-around time. Both the sensitivity and specificity exceed 97%, and for the NAP1 strain of CD approach 100%. In one large study, repeat testing with PCR of a previously negative stool within two weeks yielded a new positive result in only 2.5% of 293 patients.13
In a comparative study of 185 stool specimens at LGH, EIA proved to be only 58% sensitive compared to PCR. Discrepant samples, those where the EIA and PCR results did not agree, were forwarded to a reference lab for toxigenic culture confirmation. The toxigenic culture results matched the PCR results in 13 of 14 specimens. This new PCR methodology has recently been implemented at Lancaster General Hospital as the default diagnostic assay for CDI.
An Illustrative Case
A 53 year-old previously healthy man was admitted to LGH with abdominal pain and diarrhea. He had not been previously hospitalized, and had no connection to health care. He had undergone a root canal two weeks prior to admission, after which he was prescribed one week of oral clindamycin. An Emergency Department CBC demonstrated a WBC of 18,200 cells per cubic milliliter. Clostridium difficile toxin B gene assay by PCR was positive. Serum creatinine was normal. Initial CT scan of the abdomen demonstrated colonic wall thickening localized to the sigmoid colon. He was started on oral metronidazole and released.
He returned the following day with worsening pain, a decrease in diarrhea, and increasing weakness. WBC was now 35,000 cells/mm3. Repeat CT scan now demonstrated inflammatory involvement of the entire colon without evidence of perforation. Within 12 hours from admission developed acute renal failure, lactic acidosis, and septic shock. His WBC reached 61,800 cells/mm3. He was taken to the operating room emergently for exploration and underwent a subtotal colectomy. Postoperative WBC reached 144,000 cells/mm3. Despite aggressive supportive measures, and a second exploratory laparotomy, the patient experienced continued multi-system organ deterioration and expired on the third hospital day.
Evidence-based Treatment Guidelines
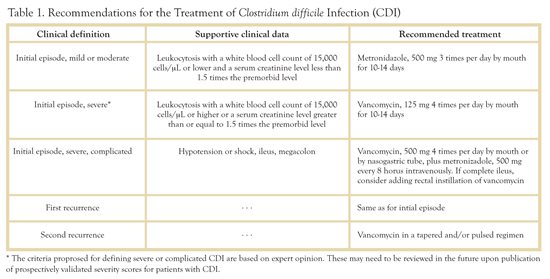
The Infectious Diseases Society of America (IDSA) and the Society for Health Care Epidemiology (SHEA) have recently published evidence-based management guidelines.14 A brief overview follows (also see Table above).
Initial Episode, Mild or Moderate Disease: This is defined as a hemodynamically stable patient with CDI and a peripheral WBC of < 15,000 and a serum creatinine of less than 1.5 times the premorbid level. For these patients, metronidazole 500 mg orally TID for 10-14 days is recommended.
Initial Episode, Severe Disease: Defined as a hemodynamically stable patient with a peripheral WBC > 15,000 or a creatinine greater than 1.5 times baseline. The recommended treatment is vancomycin 125 mg QID for 10-14 days.
Initial Episode, Severe, complicated: These are patients with fulminant colitis with hypotension, shock, ileus, or toxic megacolon. For such patients vancomycin 500mg QID by mouth or NG should be accompanied by metronidazole 500mg IV Q 8 Hrs. Vancomycin by rectal instillation may be considered in patients with ileus.
First Relapse: Treat as initial episode.
Second relapse: Vancomycin regimen in a tapered or pulsed regimen. Exact dosing schedules for treatment of the second (or subsequent) relapse remain to be elucidated by clinical studies. The rationale behind pulsed therapy is to treat with vancomycin on one day followed by several days off therapy, thus allowing residual CD spores to germinate and become therefore susceptible to antimicrobial therapy. Drugs such as cholestyramine, which bind and inactivate vancomycin, should be avoided during treatment of CDI.
Evolving New Therapies
Probiotics such as Lactobacillus acidophilus or Sachromyces boulardii are not recommended by the IDSA/SHEA guidelines due to lack of clear benefit in clinical trials. Note that vancomycin is highly active against Lactobacillus acidophilus but not other Lactobacillus species. Also S.boulardii has been associated with airborne systemic infection and fungemia in compromised hosts.15
Bacitracin administered enterally can be effective in some cases of CDI. Specific dosing regimens are empirical.16
Rifaximin is a relatively expensive drug that is not FDA-approved for the treatment of CDI, although some activity has been demonstrated. Some practitioners use this agent at a dose of 400-800mg daily in divided doses for two weeks after completion of a vancomycin course. A small study suggests efficacy as salvage therapy in patients refractory to vancomycin and metronidazole.17 However, rifamycin resistance develops rapidly.
Nitazoxanide is a drug approved for the treatment of Cryptosporidiosis. In several small, randomized studies has shown efficacy comparable, but not superior to, vancomycin and metronidazole. It is awaiting FDA approval.
Fidaxomicin (OPT-80), recently approved by the FDA, is a new macrolide antimicrobial agent that has proven in a large clinical trial to be non-inferior for the treatment of CDI. Its potential advantage lies in a lower relapse rate than vancomycin.18
Tigecycline administered parenterally, has demonstrated impressive efficacy for severe, refractory CDI in anecdotal reports.19 Further research is warranted.
Immunotherapy has been suggested as a therapeutic intervention given the protective effect of anti-CD toxin antibodies outlined above. Infusion of IVIG (Intravenous Immunoglobulin) in severe cases of toxemic CDI has been studied both in small, uncontrolled case series and small studies. Results have been inconclusive. Many practitioners, however, utilize IVIG infusion in refractory cases of CDI or in those associated with septic shock. Monoclonal antibodies to CD toxins are being researched. In addition, work has begun on the development of a vaccine for CD toxins.
Fecal Bacteriotherapy is perhaps the most controversial of the emerging therapies for CDI. This approach involves the introduction of donor stool into the GI tract of a patient with CDI in an attempt to restore normal bacterial homeostasis. A success rate of 89% has been reported in a study of 100 patients who have failed antimicrobial therapy.20 Many issues of patient safely, screening, and liability associated with this non-FDA-approved approach limit its availability.
Prevention
Given the complexities described above, it would seem obvious that an ounce of prevention is indeed worth a pound of cure. Fortunately, some specific interventions have been shown to impact the spread of CDI. Antimicrobial stewardship efforts can be utilized to curtail the use of unnecessary antibiotics, and to restrict those that might be associated with disease outbreaks. Private rooms with full barrier precautions should be implemented for all patients with CDI. Hand hygiene with antimicrobial soap and water has been shown to be superior to alcohol-based foam in the management of CDI. In addition, Diluted (1:10) sodium hypochlorite bleach should be utilized for environmental decontamination, as the spores of CD remain impervious to standard cleaning measures.21
Conclusions
CDI has re-emerged as a more virulent and more challenging infection than ever before. While the battle has been aided by new diagnostic and therapeutic modalities, clinical acumen and the sound application of evidence-based guidelines for treatment and prevention remain paramount. Ultimately, a safe and effective vaccine may be the only way to quell this burgeoning epidemic.
Resources
1. Pepin J, Valiquette L, Alary ME, et al. Clostridium difficile-associated diarrhea in a region of Quebec from 1991 to 2003: a changing pattern of disease severity. CMAJ 2004; 171:466-472
2. McDonald LC, Kilgore GE, Thompson A, et al. An epidemic of a toxin gene-variant strain of Clostridium difficile. N Engl J Med 2005; 353:2433-2441
3. DuPont, H.L. The Search for Effective Treatment of Clostridium difficile Infection. N Engl J Med 2011;364:473-75
4. O'Brien JA, Lahue BJ, Caro JJ, Davidson DM. The emerging infectious challenge of Clostridium difficile-associated disease in Massachusetts hospitals: clinical and economic consequences. Infect Control Hosp Epidemiol 2007;28:1219-27.
5. Fifth Decennial International Conference on Healthcare-Associated Infections (ICHAI) 2010:Abstract 386, presented March 20, 2010.
6. MacCannell DR, Louie TJ, Gregson DB, et al. Molecular analysis of Clostridium difficile PCR ribotype 027 isolates from Eastern and Western Canada. J Clin Microbiol 2006;44:2147-2152
7. Zilberberg MD, Tillotson GS, McDonald LC. Clostridium difficile infections among hospitalized children, United States, 1997–2006. Emerg Infect Dis. 2010;16:604–9.
8. Zilberberg, MD, et. Al. Increase in Adult Clostridium difficile-related Hospitalizations and Case-Fatality Rate, United States, 200-2005. Emerg Infect Dis. 2008 June;14(6):929-31.
9. Kelly, CP, and Lamont, JT. Clostridium difficile-More Difficult Than Ever. N Engl J Med 2008;359:1932-40.
10. Bartlett JG. Narrative review: the new epidemic of Clostridium difficile-associated enteric disease. Ann Intern Med. 2006;145(10):758.
11. Poutanen SM, Simor AE. Clostridium difficile-associated diarrhea in adults [review]. CMAJ 2004;171(1):51-8.
12. McFarland LV, Elmer GW, Surawicz CM. Breaking the cycle: treatment strategies for 163 cases of recurrent Clostridium difficile disease. Am J Gastroenterol 2002;97:1769-75.
13. Luo RF, Banaei N. Is repeat PCR needed for diagnosis of Clostridium difficile infection? J Clin Microbiol. 2010;48(10):3738.
14. Cohen, SH. El al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults: 2010 Update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31(5):000-000
15. Munoz, P el al. Saccharomyces cerevisiae fungemia: an emerging infectious disease. Clin Infect Dis. 2005 Jun 1;40(11):1625-34.
16. McFarland, LV. Alternative treatments for Clostridium difficile disease: what really works? J Med Microbiol 54 (2005), 101-111
17. Tannous, G. ei al. Therapeutic Success of Rifaximin for Clostridium difficile Infection Refractory to Metronidazole and Vancomycin. J Clinical Gastroenterol 2009;43:91
18. Louie, TJ. Et. Al. Fidaxomicin vs. Vancomycin for Clostridium difficile Infection. N Engl J Med 2011; 364:422-431
19. Herpers, BL. Et al. Intravenous tigecycline as adjunctive or alternative therapy for severe refractory Clostridium difficile infection. Clin Infect Dis. 2009 Jun 15;48(12):1732-5.
20. Bakken, JS. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe. 2009 Dec;15(6):285-9.
21. Dubberke, ER. It al . Strategies to Prevent Clostridium difficile Infections in Acute Care Hospitals. Infection control and hospital epidemiology 2008, vol. 29, supplement 1