|
Introduction
Good medical management of any disease is based on two fundamental principles: first, the diagnosis must rely on objective criteria that are sensitive and specific to the disease; and second, the diagnosis should directly dictate medical management. Yet the diagnosis, classification, and treatment of diabetes fall short of fulfilling these two basic criteria. While the diagnosis of diabetes is based on objective laboratory criteria (fasting blood glucose [FBG] > 126 or non-FBG >200 mg/dl), determination of the patient’s type of diabetes (i.e. disease etiology or mechanism of disease) is based predominantly on a set of clinical parameters such as age at time of onset and presence or absence of metabolic syndrome.1 Furthermore, such categorization provides minimal guidance about the patient’s insulin requirements. Assumptions are made about the patient’s endogenous insulin production and insulin sensitivity which can lead to suboptimal medical management.
An alternative approach would be to characterize each patient with diabetes in terms of their insulin state, which is defined by two parameters – endogenous insulin supply and degree of insulin sensitivity. As insulin state is the result of pathophysiology determined by the cause of a patient’s diabetes, characterization of their disease would thus be independent of the underlying etiology of the diabetes. Understanding a patient’s insulin state provides unambiguous information about the person’s physiologic abnormalities, and directs us to specific medical management.
Inadequacies of the Current ADA Classification System
Diabetes is a disruption of glucose metabolism due to an absolute or relative decrease in availability of insulin to the cells. The current classification of patients with diabetes was issued by the American Diabetes Association (ADA) in 2002 and uses the underlying disease etiology as its basis.2 (Table 1)
Four general categories of diabetes are recognized:
Type 1: Insulin deficiency secondary to autoimmune destruction of beta-cells.
Type 2: Diabetes due to peripheral insulin resistance. It generally presents with hyperinsulinemia at the time of diagnosis, but can progress to insulin deficiency as the disease advances.3
Type 3: This is a “catch all” category – a long list of rare and not so rare causes of diabetes including pancreatitis, hemochromatosis, Mature Onset Diabetes of Youth (monogenic diabetes), other genetic deficiencies, medications, and idiopathic causes. Type 4: Gestational diabetes.
Most patients in a general practice setting fall into the first two categories, with almost 90% considered type 2, and 10% type 1.4
There are three problems with this classification system. First, it is dependent on the putative etiology underlying the diabetes; and, except for a handful of antibody tests, there are few good objective criteria to distinguish one etiology from another. Instead, identification or classification of each type of diabetes is based on a vague set of clinical parameters which are not diagnostic, but probabilistic, simply providing clues to help categorize a patient. Thus, a child presenting with elevated blood glucose is considered to have type 1, while an adult with elevated blood glucose is type 2. This imprecise methodology can lead to inaccurate categorization of patients. With the increasing incidence of childhood obesity, an obese child with an elevated blood sugar may be type 2 or type 1. Indeed, depending on geographic location, 8-45% of children with newly diagnosed diabetes have non immune-mediated diabetes.1 To further muddy the water, many patients are presenting with clinical and biochemical features of both type 1 and type 2 diabetes.5 Further, immune-mediated diabetes can occur at any age, even in the 8th and 9th decades of life,1 and it is now believed that between 10% - 30% of adults with diabetes originally labeled as type 2, in fact have Latent Autoimmune Disease of the Adult.6
To further complicate the picture, up to 15% of children considered to have autoimmune disease do not have positive antibodies.1,7,8 While some antibodies are known to occur transiently;9 it is likely that we do not know all the antibodies that play a role in the development of diabetes. Furthermore, while autoimmune destruction is clearly the cause of beta-cell dysfunction in many people with insulin deficient diabetes, perhaps there are other, yet to be discovered, processes that cause beta-cell death.
Thus, a second limitation of the ADA classification system is that the categories are defined by what we already know about the different etiologies underlying diabetes. There is no room for a person with diabetes whose pathology is yet to be defined. If one were to create a theoretical list of all the possible ways the normal physiologic pathway of glucose metabolism could be disrupted, an enormous number of causes for diabetes could be imputed (Table 1). We know that decreased insulin production can be due to autoimmune beta-cell destruction. It can also be due to enzymatic deficiencies in the production pathway, to inflammation of the beta cells, to a deposition-type disease; it could be due to the lack of a cell receptor that turns on a beta cell; it could be due to a liver that cannot produce the appropriate feedback loops to stimulate the pancreas – or to a problem with any one of the number of stimulatory hormonal pathways/feedback loops. The presentation of diabetes may have nothing to do with pancreatic dysfunction, but may be due to a defective insulin molecule, mutations of the insulin receptor in the periphery, or defects in the post-receptor signal transduction pathway.
Some of the above pathologies are already known and well described.1 Autoimmune destruction of the beta cell has been understood for over ten years, Mature Onset of Diabetes of Youth (monogenic diabetes) are now recognized as single genetic deficiencies in the insulin production pathway.10,11
Third, the etiology-based ADA classification system is limited in its clinical usefulness because of its inability to accurately direct medical management. Diabetes, regardless of type, is a progressive disease.3,13,14 Endogenous insulin production and peripheral insulin sensitivity change with time. Early on in the disease, an individual with type 2 diabetes is hyperinsulinemic; with disease progression, they may become insulin deficient.Thus, etiology does not accurately reflect physiologic state, and unless insulin state is specifically determined, there is no way of knowing where in the disease process a patient is at any point in time. Regardless, current first line therapy for type 2 diabetes is invariably an oral hypoglycemic, with additional oral medications introduced in a stepwise fashion as the disease progresses. Which oral hypoglycemic is used first, and the order by which medications are subsequently added, is largely determined by physician preference, patient tolerance, and trial and error.3 The choice has little to do with targeting the patient’s specific deficiencies in the glucose metabolic pathway.
Multiple classes of treatment agents are now available. Each class targets a specific aspect of the glucose metabolic pathway. Metformin reduces hepatic gluconeogenisis; sulfonylureas and meglitinides increase endogenous insulin secretion; thiazolidinediones increase peripheral insulin sensitivity; alpha-glucosidase inhibitors decrease glucose absorption in the gut. Exogenous insulin obviously increases insulin supply. Recently incretins (pramlintide, exenatide), gut hormones that amplify insulin secretion and tissue response, have become available. Surely, this diversity in available treatments should allow us to tailor medical management to a specific insulin state – how much endogenous insulin the patient is producing, or their degree of insulin resistance.
The Concept of Insulin State: An Alternative Model For Defining Diabetes
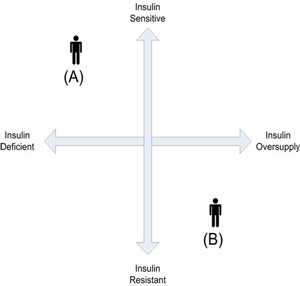
To resolve the clinical deficiencies of the ADA system, which groups patients according to disease etiology, an alternative model is proposed which characterizes patients by insulin state, a condition defined by two continuous parameters: capacity for endogenous insulin secretion, and level of insulin resistance. In the continuum for endogenous insulin production, the insulin deficient patient is at one end, and the insulin overproducer at the other. In the continuum for peripheral insulin resistance (or its reciprocal, sensitivity) the patient with insulin sensitivity is at one extreme, and the one with severe insulin resistance at the other. The two continua can be superimposed and placed at 90 degrees to each other to create four quadrants. (Figure 1). Thus, a person who is insulin deficient and insulin sensitive (the classic type 1 patient) is in the upper left corner of the upper left quadrant. A person who is over-producing insulin and is insulin resistant is in the lower right quadrant.
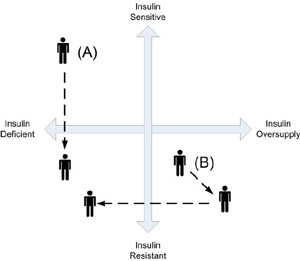
While each quadrant could be considered a representation of a certain “type” of diabetes, this interpretation is discouraged, because it is essential to this model to understand that a person’s insulin state, or diabetic phenotype, is fluid. With progression of the disease and the development of co-morbidities, both endogenous insulin production and relative peripheral sensitivity may change, and a person with diabetes may move from one quadrant to another. Thus, the classic type 1 patient with insulin deficiency and insulin sensitivity at diagnosis may not remain in the upper left quadrant. With progressive weight gain, the individual may develop increasing insulin resistance, shifting him gradually toward the lower left corner of the lower left quadrant (Figure 2 - A). The classic type 2 patient with insulin over-production and peripheral insulin resistance at diagnosis, typically becomes even more insulin resistant with progression of their disease. This increased resistance is initially met by an increase in insulin production, shifting the patient’s insulin state toward the bottom right corner of the right lower quadrant. Eventually, these increased demands on the pancreas can lead to beta-cell “burnout” and subsequent insulin deficiency, shifting the patient’s insulin state to the lower left quadrant. (Figure 2 - B). Thus, though at diagnosis patients with type 1 and type 2 diabetes have very different insulin states, with progression of disease their phenotypes may become increasingly similar, and they appear in the same quadrant.
Ideally, as stated previously, establishing a patient’s position along each of the continua should be dependent on a set of objective data. The current gold standard for assessment of beta-cell function and insulin sensitivity is the hyperinsulinemic-euglycemic clamp.12,15 However, this method is labor intensive, costly, and therefore, impractical for clinical application. Simpler surrogate indices, such as serum insulin, c-peptide, and blood glucose11,16-22 can provide information about insulin production and peripheral insulin resistance.23 Thus, a low c-peptide or serum insulin with a normal blood glucose reflects normal beta-cell function; a low c-peptide or serum insulin with a high blood glucose indicates insulin deficiency; a high c-peptide or serum insulin with a slightly elevated fasting blood glucose (e.g. 110mg/dl) implies sufficient insulin supply but relative insulin resistance; and a high c-peptide or serum insulin with a high blood glucose indicates more severe insulin resistance and relative insulin deficiency.
Caveats
Serum insulin and c-peptide measurements are often criticized for their lack of reproducibility. Oscillatory release, sensitivity to other hormonal influences, the lack of a linear relationship between glucose and insulin levels, are some of the many factors that can cause variations in measurement.11,12 Furthermore, while an elevated plasma insulin or c-peptide concentration in the presence of a high plasma glucose implies a state of insulin resistance, this concept has not been completely validated.12 However, despite these two potential shortcomings, several studies have demonstrated that these simple indices are clinically useful to characterize insulin production and peripheral insulin resistance.15,17,18,23,24,25,26
Management of Diabetes According to Insulin State
The advantage of approaching diabetes by characterization of insulin state is that it provides specific information so that medications can be selected based on the patient’s actual physiologic needs. To date, management for the classic type 2 patient (insulin over-producer secondary to peripheral insulin resistance) usually begins with one oral medication. With worsening blood glucose control over time, a second and often a third agent are added in a trial and error fashion, and insulin is usually introduced as an agent of last resort.14 However, as noted earlier, worsening blood glucose control may be the result of worsening peripheral resistance or it may be due to diminishing insulin supply. Prescribing a sulfonylurea (usually a first or second line choice by family physicians) to a patient who has failing beta cell function would not provide nearly as good results as prescribing insulin glargine. By tailoring medications to objective lab data, management can become more proactive, and glycemic control attained more rapidly. It may also save considerable expense on unnecessary prescriptions, and avoid prolonged periods of poor control with subsequent reduction in long term complications.
Conceptualizing a person’s diabetes in terms of insulin state provides numerous other advantages. First, because characterization of insulin state is based on objective data, this model eliminates the possibility of “misdiagnosing” the type and mismanaging the diabetes. Increasingly, with the availability of commercial antibody tests, adults across all age groups are being diagnosed with positive antibodies. As many as thirty percent of adults with presumed type 2 diabetes are now believed to have been misclassified, often resulting in a delay in the initiation of insulin therapy.4,6
A second advantage is that the same approach can be used in all patients with diabetes. This not only simplifies management but standardizes it, assuring proper management even if the underlying etiology of the diabetes is not known or clearly understood.
Finally, attention to insulin state accommodates progression of disease. Whenever a patient presents with worsening diabetic control, assessment of the individual’s insulin state can be made, and treatment subsequently tailored to match the new state. This approach can help dispel ambiguity about whether poor control is due to patient non-compliance or out-of-date management of a progressive disease.
Conclusion: Insulin State Should Be Used with The ADA Classification
Obviously, to promote understanding of diabetes pathology, it is important to continue to categorize diabetes in terms of etiology. At diagnosis, the patient’s type of diabetes should be established with the help of pancreatic cell antibodies and probabilistic clinical parameters such as age at time of onset. Regardless of the outcome, however, insulin state should also be established so that management can be tailored to the patient’s physiologic deficiencies, thus ensuring optimal management of each individual.
REFERENCES
- American Diabetes Association. Type 2 diabetes in Children and Adolescents. Diabetes Care 2000; 23(3): 381-389
- Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. From the American Diabetes Association Diabetes Care 2002; 25(S1): S5 – S20
- Tan MH, Baksi A, Kuahulec B, Kubalski P, Stankiewicz A, Urquhart R, Edwards G, Johns D. Comparison of Pioglitizone and Gliclazide in sustaining glycemic control over 2 years inpatients with type 2 diabetes. Diabetes Care 2005; 28(3): 544-550
- Engelgau MM, Narayan KMV, Herman WH. Screening for type 2 diabetes. Diabetes Care 2000;23(10):1563-1580
- Copeland KC, Becker D, Gottshalk M, Hale D. Type 2 diabetes in children and adolescents: risk factors, diagnosis and treatment. Clinical Diabetes 2005; 23(4):181-185
- Juneja R, Hirsch IB, Naik RG, et al. Islet cell antibodies and GAD antibodies, but not clinical phenotype help identify type 1 ½ diabetes in patients presenting with type 2 diabetes. Metabolism 2001; 50(9):1008-1013
- Nesmith JD. Type 2 diabetes in children and adolescents. Pediatrics in Review 2001; 22(5)
- Turner R, Stratton I, Horton V, Manley S, Zimmet P, Mackay IR, Shattock M Bottazzo GF, Holman R. UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. Lancet 1997; 350:1288-1293
- Niskanen LK, Tuomi T, Karjalainen J, Groop LC, Uusitupa MIJ. GAD antibodies in NIDDM. Diabetes Care 1995;18(12):1557-1565
- Winter WE, Nakamura M, House DV. Monogenic diabetes mellitus in youth. The MODY syndromes. Endo & Metab Clinics 1999;28(4):765-785
- Bergman RN, Watanabe R, Rebrin K, et al. Toward an integrated phenotype in pre-NIDDM. Diabet. Med. 1996:13(suppl 6):S67-S77.
- Matsuda M. Insulin sensitivity indices obtained from oral glucose tolerance testing. Comparison with the euglycemic insulin clamp. Diabetes Care 1999; 22(9):1462-1470
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complication in insulin-dependent diabetes mellitus. New England Journal of Medicine 1993; 329:977-986
- Bloomgarden ZT. Thiazolidinediones Diaabetes Care 2005; 28(2): 488-493
- Tripathy D, Almgre P, Tiinamaija T, Groop L. Contribution of Insulin-Stimulated Glucose Uptake and Basal Hepatic Insulin Sensitivity to Surrogate Measures of Insulin Sensitivity. Diabetes Care 2004;27(9):2204-2210
- Laakso M. How good a marker is insulin level for insulin resistance? Am J Epidemiol 1993;137:959-965
- Sluter WJ, Erkelens DW, Reitsma WD, et al. Glucose tolerance and insuli release, a mathematical approach. I. Assay of the beta-cell response after oral glucose loading. Diabetes 1976;25:241-244
- Sluter WJ, Erkelens DW, Terpstra P et al. Glucose tolerance and insulin release, a mathematical approach. II. Approximation of the peripheral insulin resistance after oral glucose loading. Diabetes 1976;25:245-249
- Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resitance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412-419
- Kahn SE, Prigeon RL, McCulloch DK, et al. Quantification fo the relationship between insulin sensitivity and beta-cell function in human subjects: evidence for a hyperbolic function. Diabetes 1993;42:1663-1672
- Turner R, Holman R, Matthews D, Hockaday T, Peto J. Insulin deficiency and insulin resistance interaction in diabetes: estimation of their relative contribution by feedback analysis from basal plasma insulin and glucose concentrations. Metabolism 1979;28:1086-1096
- Lillioja S, Mott DM, Spraul M et al. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. N Engl J Med 1993;329:1988-1992
- Hanson RL, Pratley RE, Bogardus C, et al. Evaluation of simple indices of insulin sensitivity and Insulin secretion for use in epidemiologic studies. American Journal of Epidemiology 2000; 151(2):190-198
- Tuan C, Abbasi, Lamendola C, Mclaughlin T, Reaven G. Usefulness of plasma glucose and insulin concentrations in identifying patients with insulin resistance. American Journal of Cardiology 2003;92:606 –610
- Southerland JH, Taylor GW, Offenbacher S. Diabetes and periodontal Infection: Making the connection. Clinical Diabetes 2005;23(4):171-178
-
Crownover BK, Nashelsky J. What is the best way to distinguish type 1 and 2 diabetes. J. Fam Practice 2005; 54(7):630-632
Table 1: American Diabetes Association Etiologic Classification of Diabetes Mellitus
I. Type 1 diabetes (β-cell destruction, usually leading to absolute insulin deficiency)
A. Immune mediated
B. Idiopathic
II. Type II diabetes (may range from predominantly insulin resistance with relative insulin deficiency to a predominantly secretory defect with insulin resistance)
III. Other specific types
A. Genetic defects of β-cell function: Chromosome 12, HNF-1α MODY3); Chromosome 7, glucokinase (MODY2); Chromosome 20, HNF-4α (MODY1); Mitochondrial DNA; Others
B. Genetic defects in insulin action: Type A insulin resistance; Leprechaunism; Rabson-Mendenhall syndrome; Lipoatrophic diabetes; Others
C. Diseases of exocrine prancreas: Pancreatitis; Trauma/pancreatectomy; Neoplasia; Cystic fibrosis; Hemochromatosis; Fibrocalculous pancreatopathy; Others
D. Endocrinopathies: Acromegaly; Cushing’s syndrome; Glucagonoma; Pheochromocytoma; Hyperthyroidism; Somatostatinoma; Aldosteronoma; Others
E. Drug- or chemical-induced: Vacor; Pentamidine; Nicotine acid; Glucocorticoids; Thyroid hormone; Diazoxide; β-adrenergic agonists; Thiazides; Dilantin; α-Interferon; Others
F. Infections: Congenital rubella; Cytomegalovirus; Others
G. Uncommon forms of immune-mediated diabetes: “Stiff-man” syndrome; Anti-insulin receptor antibodies; Others
H. Other genetic syndromes sometimes associated with diabetes: Down’s syndrome; Klinefelter’s syndrome; Turner’s syndrome; Wolfram’s syndrome; Friedreich’s ataxia; Huntington’s chorea; Laurence-Moon-Biedl syndrome; Myotonic dystrophy; Porphyria; Prader-Willi syndrome; Others
IV. Gestational diabetes mellitus
|
|
Figure 1: Insulin state model. A = classic type 1 patient with insulin deficiency and insulin sensitivity. B = classic type two with insulin resistance and overproduction.
|
|
Figure 2: Changing insulin state as a result of disease progression. A = increasing insulin resistance in a type 1. B = increasing insulin resistance accompanied by increase insulin production, with eventual insulin deficiency.
|
Janet Titchener Bogen, M.D.
Department of Family and Community Medicine
Clinical Advisor, Primary Health Care
The Hastings Health Centre, Hawkes Bay District Health Board
Hastings, New Zealand
|