Abstract
We examined 21 members of a Mennonite family known to be affected by pigment dispersion syndrome (PDS), a condition characterized by the release of iris pigment into the anterior chamber of the eye. Liberated pigment is usually not noticeable until adulthood and excessive release can lead to blockage of the trabecular drainage channels, glaucoma, and blindness. This study was a five-year follow-up in which we reexamined members of the first three generations as well as 11 members of the fourth generation who were not examined at the time of the original study. Clinically significant findings ranged from mild pigment release in a nineteen year old to severe end-stage glaucoma in a ninety-four year-old. The pattern of inheritance among affected individuals suggests that this family carries an autosomal dominant gene for PDS. This study has given us an opportunity to inform the family members of possible risks and to document heritable PDS in a Mennonite family. Future work with this very cooperative family might lead to the identification of the causative gene, a better understanding of the syndrome’s etiology, and more effective treatments.
Introduction
Glaucoma is a leading cause of blindness in the United States. Although clinical signs can vary, all forms of the disease are characterized by optic nerve degeneration and irreversible vision loss, usually associated with elevated intraocular pressure (IOP). The primary course of treatment involves reducing IOP by either decreasing the production of aqueous humor or increasing its outflow. Although many patients respond well to available pharmacological and surgical interventions, some have IOPs that remain high despite aggressive treatment and others exhibit progressive optic nerve degeneration despite having normal or low IOPs.
Some forms of glaucoma are inherited and studies of affected families have led to the identification of more than 20 genes.1 These recent findings support the long-held suspicion that different types of glaucoma are triggered by different underlying molecular events.2 The current understanding of these events is limited, but the identification of causative genes may lead to early detection through diagnostic genetic screening and more effective therapeutic approaches.
Pigmentary glaucoma (PG) is a form of glaucoma that is associated with pigment dispersion syndrome (PDS), a condition characterized by the liberation of iris pigment into the anterior chamber (AC). For affected North American families of western European descent, PDS appears to be an autosomal dominant trait, and one study of four families mapped a causative gene to chromosome seven (7q35-q36).3,4 The syndrome does not express itself until adulthood and does not always lead to PG.5-7 Pigment release is the result of a concave sagging in the peripheral iris which brings the posterior surface into contact with the lens zonules (Figure 1).8 Friction between the two surfaces causes pigment loss which is most apparent when viewing the anterior chamber while positioning a fiber optic transilluminator over the temporal sclera. The resulting transillumination defects form a characteristic pattern of slits in the iris that correspond to the location and radial arrangement of the zonules.
Aqueous flow carries pigment to several AC surfaces including the lens, iris, and corneal endothelium. Corneal deposits often form a centrally located band of pigment known as Krukenberg’s spindle, which may get its shape from the vertical meeting of aqueous currents (Figure 2).9 The total amount of AC pigment is apparently in flux, and showers of pigment can be triggered by pharmacological agents or vigorous exercise.10-14 Aqueous currents eventually carry the pigment granules to the trabecular drainage system where they accumulate or get phagocytized by trabecular endothelial cells.15 The exact mechanism that links liberated pigment to elevated IOP and glaucomatous optic nerve damage remains controversial, but the drainage channels of the trabecular meshwork are clearly compromised when pigment is present.9,10,16-19
Although PDS is a risk factor, longitudinal studies suggest that most affected people never develop PG.5-7 The difference between those who develop glaucoma and those who do not probably lies in the trabecular meshwork’s ability to tolerate or remove pigment.8 When accumulation is modest, trabecular endothelial cells are apparently able to phagocytize pigment and keep the drainage channels clear.15 However, excessive pigment accumulation overwhelms the clearing capacity of the endothelial cells and damages the trabecular meshwork.20 This damage can become irreversible, leading to a significant and sometimes unmanageable rise in IOP.6-9
As with primary open-angle glaucoma, the central course of treatment for PG is the reduction of IOP, but interventions should also be aimed at reducing iridozonular contact. Potash et al. used ultrasonic measurements to show that miotic therapy or laser iridotomy can eliminate the iris concavity associated with PDS.21 The normal age-related reduction in pupil size may also reduce the concavity of the peripheral iris and could explain why the symptoms of PDS and PG often decrease over time.8
The Mennonite and Amish communities of southeastern Pennsylvania offer Lancaster County healthcare providers unique opportunities to study genetic disorders.22, 23 The communities are isolated, the populations are small, and the nuclear family sizes are often large. Taken together, these factors greatly enhance the odds of isolating genes responsible for heritable diseases.23 Of course, most private practitioners do not have the technological tools to locate and identify causative genes, but by compiling information about signs and symptoms in an affected family, physicians can map pedigrees and propose modes of inheritance. This information can be shared with geneticists and used to alert family members when their risk of disease is believed to be greater than that of the general population and to support or challenge existing theories of inheritance and gene penetrance.24
This investigation is a five-year follow up of a study that identified PDS in three generations of a local Mennonite family.25 In the first investigation, we examined family members, took blood samples, and searched for a causative gene in the 7q35-q36 region of chromosome seven, the only site known to contain a PDS gene.3 Although this family shows traits consistent with an autosomal dominant pattern of inheritance, linkage analysis indicated that they do not carry the gene mapped to chromosome seven.25 Finding the causative gene will require additional genetic analyses, which will be more likely to succeed if we can identify more affected family members.
Although the original study involved over 100 family members, the current one concerns only twenty-one individuals in one branch of the family. We reexamined members of the first three generations, but our primary focus was on the 11 members of this branch’s fourth generation, most of whom were not adults (18 years or older) and therefore not examined at the time of the original study. We screened these individuals, including five children of one affected member, for early signs of PDS. Our aim was to estimate the age of onset and to determine if data from the fourth generation continue to support an autosomal dominant pattern of inheritance. The identification of additional affected individuals could justify a new search for this family’s causative gene.
Methods
After obtaining informed consent, we collected medical records and examined 21 members of one Mennonite family from Eastern Lancaster County. Each examination began with an evaluation of the anterior chamber using a slit-lamp microscope in order to search for liberated pigment, transillumination defects, and iris concavity. We measured intraocular pressures (IOPs) with a Goldman tonometer and tested visual fields with a Humphrey HFA-II perimeter (Carl Zeiss, Thornwood, New York). We also took scanning laser images of the optic nerve head topography and classified eyes as normal or glaucomatous using the Moorfields regression algorithm included in the Heidelberg Retina Tomograph II (Heidelberg Engineering, Carlsbad, California).26
Results
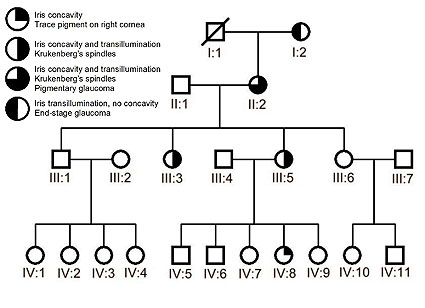
The pedigree in Figure 3 represents four generations of examined family members as well as one deceased individual (I:1) who, according to his family, had no significant visual impairments. The oldest surviving female (I:2) was ninety-four at the time of the examination and has end-stage glaucoma with significant vision loss and severe glaucomatous cupping of the optic nerves. Medical records revealed that she was diagnosed with glaucoma associated with elevated IOPs in her 50s and had cataract surgery bilaterally when she was in her mid-70s. On examination, we found bilateral transillumination defects but no pigment in the anterior chamber or trabecular meshwork. Her daughter (II:2) and two of her granddaughters (III:3 and III:5), had classic signs of PDS including bilateral Krukenberg’s spindles, concavity of the peripheral iris with transillumination defects throughout 360 degrees, and pigment deposition in the trabecular meshwork. Medical records showed that family member II:2, 75 years of age at the time of her exam, was diagnosed with PG in her early 50s. Her treatment history reveals a two-decade battle to control IOPs which often exceeded 25 mm Hg despite eyedrop therapy and surgical interventions. She currently enjoys good visual acuity, but has suffered from peripheral vision loss and progressive glaucomatous cupping of the optic nerves. Her four children in generation III, aged 48 to 52, could not provide significant ophthalmic histories from the time before the original study in May of 2000 when it was found that two of them had PDS with no evidence of glaucoma. The five-year follow-up examination revealed no detectable changes in the members of generation III; the previously diagnosed siblings, III:3 and III:5, were the only ones with PDS and all members of the generation had normal optic nerves, normal visual fields, and normal IOPs upon examination.
Unlike her siblings and cousins, one 19 year-old patient (IV:8), a daughter of affected patient III:5, exhibited trace amounts of anterior chamber pigment in the right eye and bilateral concavity of the peripheral iris. Intraocular pressures, visual fields, and optic nerve head topographies were within normal limits for this patient and all members of her generational cohort (aged 16-36).
The four individuals who married into the family (II:1, III:2, III:4 and III:7) are probably not affected since they showed no signs of PDS while in the age range (45-75 years) in which signs are most apparent.
Discussion
Without a detailed family history, the fine endothelial pigment and iris concavity in 19 year-old patient IV:8 may have been overlooked or considered clinically insignificant in the course of a routine examination. Although the anterior pigment deposition in the right eye is too fine to form a clear Krukenberg’s spindle and iris concavity is not a perfect predictor of PDS, none of the unaffected family members in generations II-IV exhibit these traits. It must be emphasized, however, that PDS is rarely detected before age 30 and other members of generation IV may develop PDS in time. The case of patient IV:8 may represent the earliest detectable stage of PDS and she has been alerted to the importance of lifelong monitoring and early intervention in order to limit the sequelae of PDS and PG. Her affected mother, aunt, grandmother, and great-grandmother did not have the benefit of such an early diagnosis, partly because none had an eye exam before age 40.
To date, patients III:3 and III:5 have no signs of elevated IOP or glaucomatous damage and may be among the estimated 80% of individuals with PDS who do not develop PG.5-7 Nonetheless, their mother has PG and they may have compromised trabecular function as a result of ongoing pigment release. Such patients should be followed closely in the event that the phagocytic capacity of the trabecular endothelial cells is overwhelmed, leading to elevated IOP and PG.19, 20 Patients II:2 and I:2 represent intermediate and late stages of PG, respectively. Despite aggressive treatment, patient II:2 has exhibited intermittent periods of unmanageable elevated IOPs for over 20 years. These episodes have been less frequent in the last five years and she has recently responded well to therapy. To date, she has not suffered significant vision loss. It is possible that, over time, age-related changes and surgical interventions may have reduced the iridozonular contact and, consequently, decreased the pigment release and obstruction of the trabecular meshwork. Patient I:2, however, represents a case of end-stage PG with significant vision loss. This is most likely due to the fact that she developed PDS and glaucoma approximately 40 years ago when the syndrome was poorly understood and treatment options were few. Ironically, as is often the case, this patient no longer exhibits Krukenberg’s spindles and the iris transillumination defects are the only evidence of past PDS. The combined effects of bilateral cataract surgery, trabeculectomies, and natural aging changes may have eventually reduced iridozonular contact enough to halt pigment liberation.8
Investigations into the causes of other types of glaucoma suggest that they are genetically heterogeneous conditions for which a specific group of clinical signs can be caused by one of several possible mutations having dominant or recessive modes of inheritance.1 The same is probably true for PDS since this family exhibits classic traits of the condition without carrying the only known gene. The pattern of inheritance described here, combined with the genetic screening conducted in the previous investigation, suggests that this family carries an unidentified autosomal dominant gene for PDS.
Conclusion
This very cooperative family has given us a unique opportunity to get a cross-sectional view of the progression from PDS to PG and, more importantly, inform the family about the risks and a possible mode of inheritance. We will continue to monitor them, especially the children of affected members, for new signs. To our knowledge, this family represents the only documented example of inherited PDS in the Mennonite community.
REFERENCES
- Wiggs JL. Genetic etiologies of glaucoma. Arch Ophthalmol. 2007 Jan;125:30-7.
- Levene RZ. Low tension glaucoma: a critical review and new material. Surv Ophthalmol. 1980 May-Jun;24(6):621-64.
- Andersen JS, Pralea AM, DelBono EA, Haines JL, Gorin MB, Schuman JS, Mattox CG, Wiggs JL. A gene responsible for the pigment dispersion syndrome maps to chromosome 7q35-q36. Arch Ophthalmol. 1997;115:384-8.
- Bovell AM, Damji KF, Dohadwala AA, Hodge WG, Allingham RR. Familial occurrence of pigment dispersion syndrome. Can J Ophthalmol. 2001;36(1):11-7.
- Mastropasqua L, Ciancaglini M, Carpineto P, Gallenga PE. Early stadiation of pigmentary dispersion syndrome and long-term analysis of progression to pigmentary glaucoma. Ann Ophthalmol Glaucoma. 1996;28:301-7.
- Sugar HS. Pigmentary glaucoma: A 25 year review. Am J Ophthalmol. 1966;62(3):499-507.
- Migliazzo CV, Shaffer RN, Nykin R, Magee S. Long-term analysis of pigmentary dispersion syndrome and pigmentary glaucoma. Ophthalmology. 1986;93(12):1528-36.
- Campbell DG. Pigmentary dispersion and glaucoma. A new theory. Arch Ophthalmol. 1979;97(9):1667-72.
- Richardson TM. Pigmentary dispersion syndrome and glaucoma. In Albert DM, Jakobiec FA (eds.). Principles and Practice of Ophthalmology. W.B. Saunders Co., Philadelphia, 1994, pp.1414-25.
- Kristensen P. Mydriasis-induced pigment liberation in the anterior chamber associated with acute rise in intraocular pressure in open-angle glaucoma. Acta Ophthalmol (Copenh). 1965;43(5):714-24.
- Epstein DL, Boger WP 3rd, Grant WM. Phenylephrine provocative testing in the pigmentary dispersion syndrome. Am J Ophthalmol. 1978;85(1):43-50.
- Haddad R, Strasser G, Heilig P, Jurecka W. Decompensation of chronic open-angle glaucoma following mydriasis-induced pigmentary dispersion into the aqueous humour: a light and electron microscopic study. Br J Ophthalmol. 1981;65(4):252-7.
- Mapstone R. Pigment release. Br J Ophthalmol. 1981;65(4):258-63.
- Haynes WL, Johnson AT, Alward WL. Effects of jogging exercise on patients with the pigmentary dispersion syndrome and pigmentary glaucoma. Ophthalmology. 1992;99(7):1096-103.
- Sherwood M, Richardson TM. Evidence for in vivo phagocytosis by trabecular endothelial cells. Invest Ophthalmol Vis Sci. 1980;19(4 suppl):66.
- Grant WM. Experimental aqueous perfusion in enucleated human eyes. Arch Ophthalmol. 1963;69:783.
- Murphy CG, Johnson, Alvarado JA. Juxtacanalicular tissue in Pigmentary and primary open angle glaucoma. The hydrodynamic role of pigment and other constituents. Arch Ophthalmol. 1992;110(12):1779-85.
- Campbell DG, Schertzer RM. Pathophysiology of pigment dispersion and pigmentary glaucoma. Curr Opin Ophthalmol. 1995;6(2):96-101.
- Gottanka J, Johnson DH, Grehn F, Lutjen-Drecoll E. Histologic findings in pigment dispersion syndrome and pigmentary glaucoma. J Glaucoma. 2006;15(2):142-51.
- Richardson TM, Hutchinson BT, Grant WM. The outflow tract in pigmentary glaucoma: a light and electron microscopic study. Arch Ophthalmol. 1977;95(6):1015-25.
- Potash SD, Tello C, Liebmann J, Ritch R. Ultrasound biomicroscopy in pigment dispersion syndrome. Ophthalmology. 1994;101(2):332-9.
- Morton DH, Morton CS, Strauss KA, Robinson DL, Puffenberger EG, Hendrickson C, Kelley RI. Pediatric medicine and the genetic disorders of the Amish and Mennonite people of Pennsylvania. Am J Med Genet C Semin Med Genet. 2003;121(1):5-17.
- Puffenberger EG. Genetic heritage of the Old Order Mennonites of southeastern Pennsylvania. Am J Med Genet C Semin Med Genet. 2003;121(1):18-31.
- Qureshi N, Bethea J, Modell B, Brennan P, Papageorgiou A, Raeburn S, Hapgood R, Modell M. Collecting genetic information in primary care: evaluating a new family history tool. Fam Pract. 2005;22(6):663-9.
- Gallagher SP, Lehmann OJ, Leonardo D, Ebenezer N, Ocaka L, Child A, Hitchings RA, Sarfarazi M, Hattacharya SS. Linkage analysis of a large Amish pedigree with glaucoma. Invest Ophthalmol Vis Sci. 2001;42(4 suppl):3020.
- Wollstein G, Garway-Heath DF, Hitchings RA. Identification of early glaucoma cases with the scanning laser ophthalmoscope. Ophthalmology. 1998;105(8):1557-63.
Figure 1. Slit lamp photograph of an eye with significant concavity in the peripheral iris. A vertical slit of light is cast through the cornea and upon the inferior iris. The superimposed oval represents the pupil’s perimeter and the arrow indicates the point of greatest concavity, where the posterior iris is most likely to be in contact with the lens zonules.
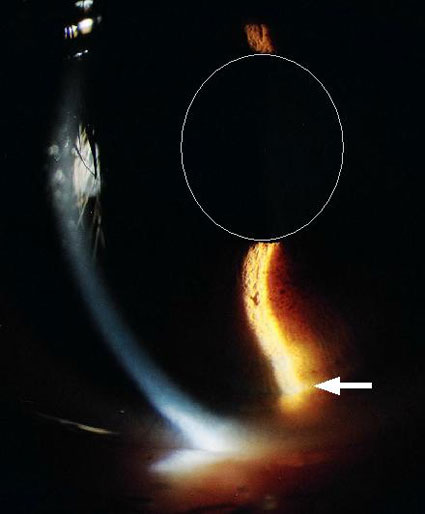
Figure 2. Slit lamp photograph showing a dense vertical band of pigment forming Krukenberg’s spindle on the corneal endothelium. This patient is the mother of the patient in Figure 1.
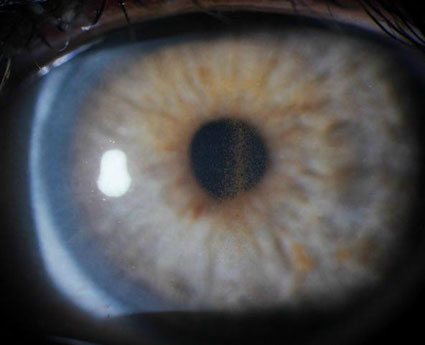
Figure 3. The family pedigree reveals a pattern of inheritance consistent with an autosomal dominant PDS gene.
Special Reviewer’s Comment by D. Holmes Morton, M.D.
This paper is a nice example of Translation Genetics within a local medical system. It explicitly addresses a significant problem within an extended family, and demonstrates how often the medical care required to be helpful is quite basic. The ingredients are: a good history, good follow-up, and techniques of monitoring that are readily available - including linkage analysis to search for the gene locus and to help better define the risk of disabling disease. This kind of work should be done more often, both in Lancaster County and in University Hospitals.
The marker studies done in this paper looked for linkage to specific clusters of genes within chromosome 7 and 18 that were previously reported to be associated with PDS and glaucoma, but data indicate the causative gene is elsewhere. With the same DNA collected for this paper, the authors could redo the analysis in subsequent studies, using 10,000-50,000 SNP* markers to search the entire genome for the disease gene. The goal of such studies is to find the specific gene and the mutation in that gene which gives rise to the PDS. Detection of the mutation by a polymerase chain reaction (PCR) assay then becomes a simple and inexpensive diagnostic test.
At the Clinic for Special Children we have a catalogue of 105 genetic syndromes found in our patient population. For 84 of these disorders we currently know the gene locus, and for 75 of the disorders we know the specific disease-causing mutation. SNP mapping studies are constantly being done to find the gene locus and mutation for the other 21 syndromes. This summer, with support from Franklin and Marshall College and Lancaster General Hospital, and with the help of Mike Fox, a 2007 F&M Graduate, we are developing a new method to detect specific gene mutations. The method will allow 96-380 samples to be analyzed in approximately 2 hours for a cost of less than $10 per sample, which will make it practical to carry out rapid detection of mutations in high-risk newborns, and testing for carriers within large extended families.
Many adult-onset genetic disorders will - like PDS – also be dominant traits with variable penetrance. Such mutations may be important risk factors for disease, but they seldom act alone. Expression of disease is the product of interacting genes which control proteins, and other factors such as biochemistries and environmental influences. In patients with PDS, there might be influences from factors such as infections that cause inflammation of the eye; injuries that affect microanatomy; and the availability of nutrients like vitamin C and copper, which are necessary for the formation of stable forms of the connective tissues that form the microscopic drainage pathways of the anterior chamber, and support the iris and lens.
The first evidence that a disease is treatable is often the variation in the natural history of the disease. Preventive treatments arise from understanding how the interaction of so many variables influences the disease. An effective approach might include attention to many aspects: treating infections early, suppressing inflammation, avoiding injury, taking a multivitamin, and using medication to regulate interacting genes up or down. The latter is the insight that is most closely related to my own work on the influence of genes on human biology.
*A
Single Nucleotide Polymorphism (SNP) is an inherited variation in a single base-pair of DNA. In contrast to a disease causing mutation, a Polymorphism does not cause loss of function of a Gene. Most SNPs are found in Introns, which are the non-coding regions of DNA between the coding regions of a Gene, called Exons. The SNPs on the 10,000 and 50,000 SNP genotype arrays used at the Clinic are in non-coding regions of DNA; newer genotype arrays identify 1 - 2 million SNPs and include Polymorphisms in Exons. There are an estimated 5-10 million SNPs in the human genome. The precise location of each SNP can be found in the Human Genome Databases on-line at the National Center for Biotechnology Information.
Shawn P. Gallagher, Ph.D.
Assistant Professor of Psychology
Department of Psychology, Millersville University
103 Byerly Hall
Millersville, PA 17551
Phone – 717-971-2354
sgallagher@millersville.edu
Barton L. Halpern, M.D., F.A.C.S.
Eye Doctors of Lancaster
140 North Pointe Boulevard
Lancaster, PA 17601
Phone – 717-560-4020
Fax – 717-560-2919
bhalpern@aol.com