Winter 2022 - Vol. 17, No. 3
SCIENTIFIC REPORT
A Woman Cured of Refractory Atypical HUS-TTP
after Bilateral Nephrology
L. Dumasia S. Dumasia Rodenberger
Lena Dumasia, MD
Hemotologist/Oncologist, Lancaster Cancer Center
Suraj Dumasia
Student, Manheim Township High School
Charles Rodenberger, MD
Nephrologist, Hypertension and Kidney Specialists
INTRODUCTION
This case report describes a patient who was diagnosed with atypical hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) who required continuous plasma exchange for a period of eight months. She developed seizures when attempts were made to wean her off plasma exchange, and her case was not responsive to immunosuppression and treatment with rituximab (Rituxan). Ultimately, however, her microangiopathy resolved after she underwent a bilateral nephrectomy.
CASE REPORT
A 24-year-old female was in her usual state of wellness until she presented in April 2008 to Lancaster General Hospital (LGH) with profound fatigue. Her exam was normal, including no evidence of rash; vitals were also normal, including a normal blood pressure. Lab evaluation revealed
de novo acute renal failure (Cr = 13 mg/dL, ref. 0.5-1.0 mg/ dL), with microangiopathic hemolytic anemia — hemoglobin = 6.7 g/dl (ref. 12-16 g/dl) — and thrombocytopenia — platelet count = 96,000 platelets/
μL (ref. 150,000-450,000 platelets/
μL). Her lactic acid dehydrogenase (LDH) was elevated to 284 U/L (ref. 48-115 U/L) on presentation. Her urinalysis was consistent with acute tubular nephritis. As reference, labs were drawn two years earlier revealing a creatinine of 0.8 mg/dL and a hemoglobin of 11.1 g/dL.
Follow-up labs conducted at that time revealed that her ANA (antinuclear antibodies) was negative and a haptoglobin was <5.8 mg/dL (ref. 41-165 mg/dL). A stool study for
E. coli 0157:H7 was also negative, and a blood smear revealed only a scant amount of schistocytes. However, an ADAMTS-13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) test was normal.
1 The conclusion by her care team at LGH was that she had an atypical HUS-TTP.
After several days of dialysis and blood product resuscitation, she underwent a percutaneous renal biopsy.
This revealed 13 to 16 glomeruli, found to be globally sclerotic. Three glomeruli revealed significant evidence of thrombotic microangiopathy, and she was positive for perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA). The biopsy demonstrated a cellular crescent, presumed to be consistent with an anti-myeloperoxidase (MPO) vasculitis (see Figs. 1a and 1b). Thus, atypical HUS-TTP remained the working diagnosis.
 Fig. 1a. 20x magnification, showing thyroidization of the kidney tissue, including dilation of tubules in the upper left-hand corner, chronic change features usually seen in end-stage renal disease.
Fig. 1a. 20x magnification, showing thyroidization of the kidney tissue, including dilation of tubules in the upper left-hand corner, chronic change features usually seen in end-stage renal disease.
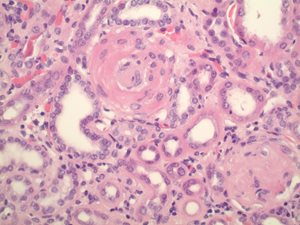
Fig. 1b. 40x magnification, demonstrating an atherosclerotic small blood vessel in the upper center and a sclerotic glomerulus in the lower right-hand corner. These features are also consistent with chronic kidney disease, most often seen in those much older than this patient.
At the time of her admission, the patient was treated with daily plasma exchange, as well as hemodialysis three times a week. Her platelets and hemoglobin stabilized, then rose at the time of hospital discharge (see Fig. 2).
 Fig. 2. Patient’s LDH and platelet count from first admission through postop. Normal range for LDH = 49-206 U/L and for platelet count = 150-450 *10^3/μL.
Fig. 2. Patient’s LDH and platelet count from first admission through postop. Normal range for LDH = 49-206 U/L and for platelet count = 150-450 *10^3/μL.
In the setting of a positive ANCA and MPO, she was placed on daily prednisone and mycophenolate mofetil (CellCept). She was discharged from LGH on May 6, 2008. She continued hemodialysis three times per week for end-stage renal disease; daily plasma exchange was continued after her discharge home.
When her LDH levels fell, she was weaned off plasma exchange. However, her LDH began to rise again, and she developed a new-onset headache on May 16, 2008, thus she was readmitted to LGH, had a witnessed seizure, and was subsequently started on levetiracetam (Keppra).
She reinitiated daily plasma exchange and required twice-daily plasma exchange treatments. During this admission, her LDH peaked at over 400 U/L. Her platelets nadired to a low of 85,000/
μL. She appeared to clinically respond to prednisone, CellCept, and plasma exchange. However, despite this initial treatment, she continued to have headaches and nausea, and her platelets began to decline again. After a discussion with the patient and her mother, a decision was made to transfer her to a tertiary care academic center for additional evaluation and management. She was transferred to Johns Hopkins University Hospital on June 6, 2008.
On the morning of June 9, 2008, the patient demonstrated an altered mental status. She would not speak in response to questions and would only follow simple commands. There was a concern for stroke, metabolic derangement, or seizure. A head CT was negative, and an EEG showed no focal seizure activity but did demonstrate severe diffuse cerebral disturbance consistent with metabolic derangement. This was attributed to uremia, and a consult was placed to the neurology service. The hematology service and rheumatology service were also involved in the workup.
The patient’s platelets at this time ranged between 80,000-120,000/
μL, and her LDH was in the 300s. Plasmapheresis was initially started but then held by discharge. She required a transfusion of two units of packed red blood cells when her hemoglobin dropped to 6.3 g/dl. ADAMTS-13 was again negative, and consulting providers thus concluded that TTP was unlikely.
1 Additionally, ANCA was negative, and ANCA-associated vasculitis did not adequately explain the clinical picture.
Systemic lupus erythematosus, immunoglobulin A nephrology, microscopic polyangiitis, and antiphospholipid syndrome were felt to be possible, but unlikely, as their findings were inconsistent with the full clinical picture. Her providers also believed that she did not have a rheumatologic process. The hematology department considered as their final diagnosis that renal disease plus HUS could be explained by Factor H or Factor I deficiency.
A lack of adequate red cell production as evidenced by anemia and persistently low reticulocyte count led to bone marrow biopsy, however this was negative for any conclusive pathologic hematological process. There was no indication for immunosuppression, and consulting physicians agreed there was no evidence of vasculitis after repeat studies. It was not possible to identify any precipitant (e.g., quinine) for her disease. She was discharged home from Johns Hopkins on June 17, 2008.
The patient was weaned off her seizure medications at the time of discharge. As the medical team believed that TTP was unlikely, the patient was also weaned off treatment with plasma exchange as she was discharged from Johns Hopkins. Unfortunately, she suffered another seizure while at home in Lancaster and was readmitted to LGH in July 2008. At that time, her LGH care team resumed her anti-seizure medications and plasma exchange treatments in accordance with the original working diagnosis of atypical HUS-TTP.
She then stabilized clinically with twice-weekly plasma exchange treatments. When attempts were made to decrease her plasma exchange treatments to once weekly, her platelet count declined from 155 to 94 to 85 platelets/
μL; concurrently, her LDH rose from 186 U/L to 221 U/L to 246 U/L. Therefore, she was maintained on plasma exchange twice a week.
2
The patient began treatment with peritoneal dialysis and was placed on the kidney transplant list. In October 2008, treatment with Rituxan 375 mg/m2
3 weekly along with twice-weekly plasma exchange
2 did not result in any meaningful improvement in her platelet count.
As it was impossible to wean her off plasma exchange after five months of twice-weekly plasma exchange treatments, an outside consultation was obtained from James George, MD, professor of medicine at the University of Oklahoma, former president of the American Society of Hematology, and a nationally known expert in platelet disorders. He reasoned that the kidneys were the source of her atypical HUS-TTP and thus recommended a bilateral nephrectomy as the curative treatment for her ongoing hemolysis.
4,5
The patient ultimately underwent this operative procedure on December 23, 2008. Pathology of the bilateral nephrectomy was consistent with Morcellated kidneys showing end-stage changes. Renal artery margins showed moderate intimal thickening with no evidence of vasculitis of either the renal artery or renal vein.
Within a week of her nephrectomy, the patient’s LDH normalized, her platelet count rebounded to normal levels, and her hemolysis completely resolved. In a short time, she no longer needed plasma exchange treatments. She remained on dialysis for several years after this cure and did well without any further requirements for plasma exchange. She was awaiting a kidney transplant when she died suddenly in the setting of substance abuse, complicated by concurrent dialysis treatment nonadherence.
DISCUSSION
In this case, despite eight months of therapy with plasma exchange, immunosuppression, and Rituxan treatments, the patient was unable to be weaned off plasma exchange. The nidus of her microangiopathic hemolytic anemia appears to have been her renal disease. When the kidneys were removed, her hemolysis resolved.
The efficacy of plasma exchange as a cure in atypical HUS-TTP remains around 75%.
6 The use of plasma exchange is considered successful when the platelet count remains >150K over a minimum period of two days.
7 There remain resistant and/or refractory cases in which the use of Rituxan, steroids, or the more recently approved Caplacizumab may also be utilized to cure the disease.
8 However, some cases may remain refractory to all of the above, such as was true this patient.
4,5
Very limited data indicate that bilateral nephrectomy may serve as a rescue therapy in plasma-exchange resistant disease due to the assumption that the kidney microvasculature is the site of platelet consumption and the ongoing nidus of disease persistence.
1,2 In patients with atypical HUS-TTP who present with end-stage renal disease refractory to plasma exchange and other accepted therapies, bilateral nephrectomy may be considered as a potential curative option.
1,2
ACKNOWLEDGEMENT
Special thanks to Bruce E. King, MD, who helped prepare and describe the specimens in Figs. 1a and 1b.
REFERENCES
1. Vesely SK, George JN, Lämmle B, et al. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients.
Blood. 2003;102(1):60-68.
2. Nguyen L, Li X, Duvall D, et al. Twice-daily plasma exchange for patients with refractory thrombotic thrombocytopenic purpura: the experience of the Oklahoma Registry, 1989 through 2006.
Transfusion. 2008;48(2):349-357.
3. Lim W, Vesely SK, George JN. The role of rituximab in the management of patients with acquired thrombotic thrombocytopenic purpura.
Blood. 2015;125(10):1526-1531.
4. Feldman M, Bruno T, George, J, et al. Bilateral nephrectomy for treatment resistant systemic lupus erythematosus and thrombotic thrombocytopenic purpura: a case report.
Am J Hematol. 2007;82(6):496-497.
5. Remuzzi G, Galbusera M, Ruggenenti P, et al. Bilateral nephrectomy stopped disease progression in plasma-resistant hemolytic uremic syndrome with neurological signs and coma.
Kidney Int. 1996;49(1):282-286.
6. Pan Z, Zhang Z, Yang Y, Hao W. The efficacy and safety of plasma exchange in the treatment of thrombotic thrombocytopenic purpura.
J Healthc Eng. 2022;2022:3519937.
7. Maloney N, Martin I, Szczepiorkowski ZM, Dunbar NM. Therapeutic plasma exchange for thrombotic thrombocytopenic purpura with refractory thrombocytopenia.
J Clin Apher. 2018;33(3):436-438.
8. Cuker A, Cataland SR, Coppo P, et al. Redefining outcomes in immune TTP: an international working group consensus report.
Blood. 2021;137(14):1855-1861.